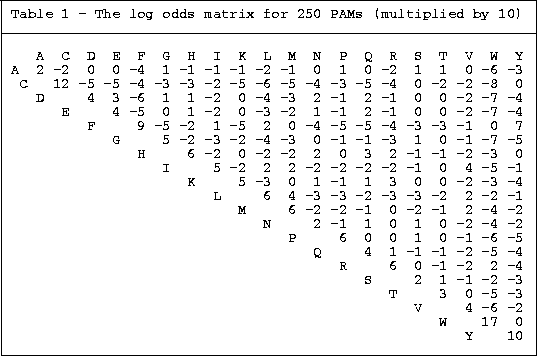
In sequence comparison, costs or similarities were previously determined by simple scoring methods such as Hamming distance, where the basic considerations are match or mismatch.
In biological macromolecules, residues may be mismatched, but still similar e.g. in chemical nature, and mismatch may not be as crucial for function.
There are also many examples where dissimilar substitutions in sequences from different species are tolerated with no loss of function.
More sophisticated methods for measuring cost or similarity have therefore been designed.
1. PAM (Percent Accepted Mutation) Matrices
Basic Construction Steps:
Therefore used to find more highly diverged sequences in
sequence homology searches.
PROBLEM 1.
For the previous problem on Uracil-DNA glycosylase from
Shope papilloma virus (SWISS-PROT Database Accession number P32941) search
for orthologs use MPsrch, varying PAM values from 100 to 400.
Resources: VSNS course Chapter
1.3.
PAM matrices
in Bioinformatics course at McMaster University
2. BLOSUM
Matrices
Derived from the BLOCKS database of conserved sequences
from protein families.
BLOSUM 62: the matrix is calculated so that sequences
more than 62% identical are merged, so that the contributions of multiple
entries of closely related sequences is avoided. Most closely related
to the PAM 160 matrix.
BLOSUM matrices may be
more useful for heuristic methods such as FASTA and BLAST, less so for others.
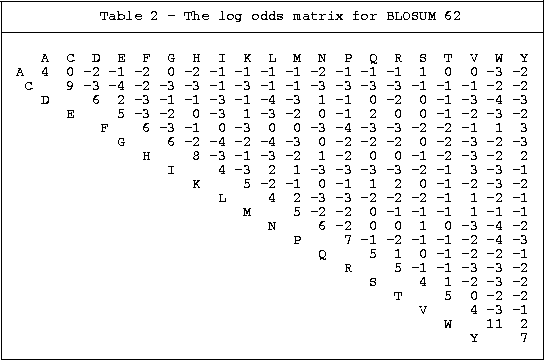
Compared to the PAM 160 the BLOSUM 62 matrix is less tolerant of substitutions
to or from hydrophilic amino acids, while more tolerant of hydrophobic changes
and of cysteine and tryptophan mismatches.
Resources:
BLOSUM
Matrices (McMaster University).