

PROTEIN STRUCTURE AND STRUCTURAL GENOMICS
TERTIARY PROTEIN STRUCTURE
Primary repository for protein structures - Protein Data
Bank (PDB)
EXAMPLE:
Get a list of the lipocalins (odorant binding proteins)
in PDB
i) by searching PDB
ii) by a BLAST search with Bovine
Odorant Binding Protein (P07435) restricting
the output to the PDB database
Get the crystal unit cell dimensions and 3-D structure
of the Bovine Odorant Binding Protein
1PBO
Protein structures may also be accessed via Entrez Structure
Get a list of Odorant-binding proteins from Entrez Structure,
and the alternate and related 3D domains of P07435
(Use "Links" links)
Using the Molecular Modelling Database (MMDB):
i) Find the
principal domains of 1PBO and a multiple alignment to related domains
ii) View the 3-D structure using Cn3D (download required)
iii) View the 3-D overlapped structures of 1PBO
with the related structures 1RLB and
1BSO
using VAST with Cn3D or Swiss-PDB Viewer
(Get Structure Neighbors from the main MMDB Structure
Summary site)
CLASSIFICATION OF PROTEIN STRUCTURES
Protein structures are classified on the basis of folds
- structural combinations of 2-D elements
DATABASES OF PROTEIN FOLDS
SCOP -
a comprehensive description of protein structures and evolutionary relationships
based on a hierarchical classification scheme
CATH
- a hierarchical classification system describing all known protein domain
structures
Proteins clustered at four levels: C(lass) - alpha, alpha
and beta, or beta
A(rchitecture) - describes domain shape
T(opology) - describes fold families, and
H(omologous superfamily) - clusters proteins likely to
share homology, have common ancestor
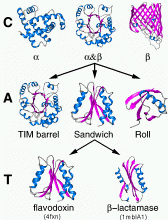
EXAMPLE
Find the domains of the BOP, PDB id 1pbo, from CATH
Get its Architecture, Topology and Homologous superfamily
members, and further information from PDBSum
Protein 3-D Structure Prediction Methods
Stanford Computational Biology Course:
Lectures Nov 3 slides 25 - 43, and Nov 24, 1999 (Protein
Folds and Protein Structure Superposition)
Fold recognition lecture and examples in Ph.D. course at Denmark Technical University
Prediction from Sequence - the CASP Competitions
- the "Holy Grail" for the structural biology community.
- subject of a biennial competition
and meeting (CASP). Proteins whose structures are likely to be
solved by NMR or X-ray crystallography
by the meeting are provided in advance to researchers, who
are then challenged to predict
3-D structures by the meeting.
Summary of CASP3: Koehl, P. and Levitt, M. (1999) Nature Structural Biology 6 (2), 108-111.
of CASP5: PROTEINS: Supplement
6, 2003
Categories of Structure Prediction:
1. Comparative modelling using a known template structure
Attempts to build a structural model
on the basis of close sequence similarity (detectable by FASTA or PSI-BLAST)
to a template protein of known structure.
Even possible for sequence similarity below 25% with use
of multiple sequence alignments, alignment of actual and predicted secondary
structures (PHD, using neural networks, most popular) and manual adjustments.
CASP3: best current methods can build a reasonable model
when when sequence similarity is significant (chance of a false match <
0.01).
2. Fold recognition
- also known as threading or 3- dimensional profile matching
- use structure to assess the compatibility
of the target sequence with each member of a library of
known protein folds e.g. SCOP.
- in CASP3 17 targets could be predicted
by this method even though none could have been recognized
by the best sequence comparison
methods such as FASTA or PSI-BLAST.
- the easiest model in this category is as good as the most difficult model in the previous category
3. Ab initio prediction
-attempts to build a model for the target protein without using a specific templatestructure
approaches include:
(i) using secondary structure, using sequence fragments & various refining methods
(ii) minimizing a physical potential energy function using a simplified protein representation
(iii) using a simplified protein model with some predictions of turns from secondary structure
Overall conclusions & questions
(i) prediction problem still very difficult but area advancing
(ii) is it more important to model a few sequences very well or many fairly well?
4. Application of Genetic Algorithms
A limited set of predicted approximate 3D structures is
constructed which meet a preassigned fitness function (e.g. Root Mean Square
Deviation of atomic coordinates). Conformational parameters (e.g. torsional
angles between atomic bonds) of these structures are then changed to a small
degree and the new set of structures assessed to determine if new structures
have been generated that better meet the fitness function (i.e. have lower
RMSD values in this case). Inferior structures are rejected from the
set and the procedure repeated a specified number of times.
5. Probable surface similarity comparison -
1. Consurf:
enables the identification of functionally important regions on the surface
of a protein or domain, of known three-dimensional (3D) structure, based
on the phylogenetic relations between its close sequence homologues.
2. Database of
proteins with WD repeats, including search tool, links to other resources.
The WD-repeat proteins are found in all eukaryotes and
are implicated in a wide variety of crucial functions. The solution of the
three-dimensional structure of one WD-repeat protein and the assumption that
the structure will be common to all members of this family has allowed subfamilies
of WD-repeat proteins to be defined on the basis of probable surface similarity.
Proteins that have very similar surfaces are likely
to have common binding partners and similar functions.
RESOURCES
Fold Databases
On-line biological services for protein structure prediction
Protein fold recognition servers
PSIPRED. including GenTHREADER at University College
London
PredictProtein
server at EMBL
UCLA-DOE protein fold recognition server
EXAMPLE:
Predict the fold structure of the protein of sequence
below:
MTSSQFDCQYCTSSLIGKKYVLKDDNLYCISCYDRIFSNYCEQCKEPIESDSKDLCYKNRHWHEGCFRCN
KCHHSLVEKPFVAKDDRLLCTDCYSNECSSKCFHCKRTIMPGSRKMEFKGNYWHETCFVCEHCRQPIGTK
PLISKESGNYCVPCFEKEFAHYCNFCKKVITSGGITFRDQIWHKECFLCSGCRKELYEEAFMSKDDFPFC
LDCYNHLYAKKCAACTKPITGLRGAKFICFQDRQWHSECFNCGKCSVSLVGEGFLTHNMEILCRKCGSGA
DTDA
(PredictProtein gives the following result:)
Post translational modifications