Motif Identification in biological sequences is valuable for:
1. eMOTIF at Stanford University
A motif is expressed as a pattern specifying the amino acids that can occur at each position in a sequence.
The possibilities are:
`.', which allows any amino acid,
a specific amino acid, or
a group of amino acids that share
some physico-chemical property, e.g. charge, hydrophobicity, size
EMOTIF ranks the motifs that it finds by both their specificity (expected false positives) and the number of supplied sequences that it covers (true positives). The twenty highest-scoring motifs are returned, and can be used to search the entire SWISS-PROT database.
Resource: eMOTIF and related resources.
2. SPLASH - Structural Pattern Localization Analysis by Sequential Histogram (requires installation).
3. TEIRESIAS
- identifies and highlights motifs common to several nucleotide or protein
sequences.
4. Cluster - Buster: identifies clusters
of user - specified motifs.
5. SIRW - combines sequence motif searches with keyword searches.
6. MinimotifMiner:
analyzes protein queries for the presence of short functional motifs
that, in at least one protein, has been demonstrated to be involved in posttranslational
modifications, binding to other proteins, nucleic acids, or small molecules,
or proteins trafficking.
EXAMPLE:
Find the proteins with nuclear receptor activity that
contain the motif LLxxL in the SwissProt database.
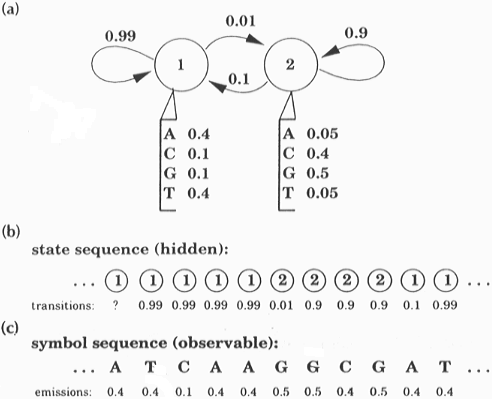
HIDDEN MARKOW MODELS
Models related to profiles, which describe multiple alignments in terms of states and sets of probabilities.
Based on the concept of Markov chain:
The states refer to each position in the alignment and are of three types:
1. Main (or match) states: contain the probabilities for each residue (e.g. 0.5A, 0.3C, 0.1G, 0.1T)
2. Deletion states: contain no residue (i.e. refer to a deletion)
3. Insert states: contain inserted residues after the corresponding main states (i.e. refer to insertions).
4. Start and End states
The probabilities are:
1. The emission probability - the probability that residues A,B, etc will occur at a given position
2. The transition probability - the probability that one state (see above) will change into another.
Insert states have an internal loop to allow for insertion of multiple residues.
In constructing HMMs, emission probabilities are generally not allowed to be zero, to allow for certain residues to occur in larger sequence sets, even though they do not occur at a given position in the sequence set under study. Probabilities are calculated as Dirichlet Priors from the Dirichlet distribution. An analogy is calculation of the probability that e.g. a 5 will be thrown next in a pair of weighted dice, even though a 5 has not been thrown in the last four throws of the dice.
The probability structure for an aligned set of sequences can then be used to identify other sequences (e.g. in a database) with the same sets of probabilities, similar primary structure and hence similar biological activity.
In HiddenMMs the states are hidden (just sets of probabilities), but can generate visible symbols.
Once a HMM has been constructed for a sequence family, other motifs or sequences which conform to that model may be identified by database searching.
Steps in use of HMMs to detect motifs in unknown sequences or remote homologies:
1. Create a multiple alignment from a family of related sequences
2. Calculate corresponding probabilities and construct a HMM (automatically)
3. Search a sequence database with the HMM (automatically)
The simplest form of HMM is the linear type, but more complex
models can also be constructed, e.g.
OTHER USES OF HMMS:
1. Ab initio gene finding
2. Radiation hybrid mapping
3. Secondary and tertiary protein structure prediction
4. Detection of unequal evolutionary
rates in molecular sequences
EXAMPLES:
PDGF protein family: see Hidden Models in Biopolymers. Science (1998) 282: pp 1436-7
GC-rich DNA sequence tracts: BCG lecture at Weizmann Institute
HMM TOOL PACKAGE
EXAMPLE: provided at site above
PACKAGES REQUIRING Download or subscription
HmmBuild and HmmSearch in Celera Discovery System
HMMer at Washington University. Also enables searches of the Pfam database of protein motifs and HMMs
SAM at UC Santa Cruz
HMMPro (commercial)
- nifty graphics & additional tools
OTHER RELATED RESOURCES
Computational Biochemistry Course at Stanford Lectures:
Pattern Matches and Consensus Sequences
Blocks, Profiles & Hidden Markov Models
Protein Profile vs Protein or DNA Database; Sequence vs
Profile database searches on
Decypher (link at introduction to above lecture )
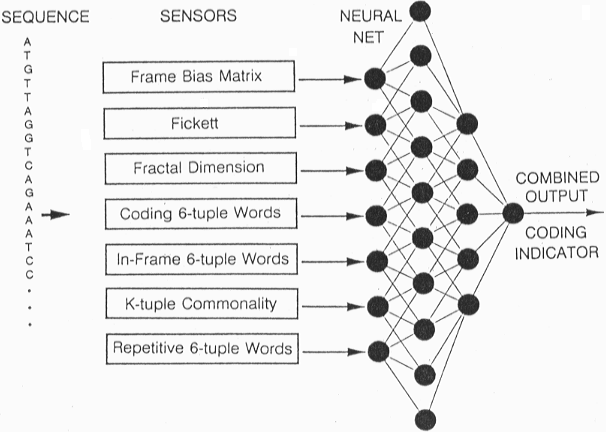
3. NEURAL NETWORKS
Models consisting of networks with interconnected units
evolving in time. Most current applications involve Layered Feed-forward
or Multilayer Perceptron (MLP) Architechture with hidden and
visible layers. Visible layers are in contact with the outside
world, and include Input and Output layers.
Output layer - expresses structural or functional features
|
Hidden layer(s)
|
Input layer - where e.g. sequence data is encoded
As with HMMs, models are initially generated and trained on existing data set, then used to predict sequence and structure information on query sequences or structures.
Applications in pattern recognition include motifs, alpha helices, splice sites, exons.
Example: GRAIL method for prediction of coding sequences:
(see BCG course for more information)
4. GIBBS SAMPLING METHOD
Example: Identification of sequences mediating coordinate transcriptional regulation in S. Cerevisiae
Motifs in 5' upstream regions identified with AlignAcein Church lab. at Harvard Med. School
(Publication available online).
5. OTHER METHODS
MOTIF AND FINGERPRINT DATABASES
BLOCKS - conserved sequence blocks in protein families
InterPro Integrated Resource of Protein Domains and Functional Sites
Pfam - Protein families database of alignments and HMMs
PRINTS - database of protein fingerprints (group of conserved motifs)
ProDom - protein domain database
PROSITE - database of protein families and domains
SMART - domain identification
EXAMPLE:
Find the domains and homologs of the protein:
MNSSSANITYASRKRRKPVQKTVKPIPAEGIKSNPSKRHRDRLNTELDRLASLLPFPQDVINKLDKLSVL
RLSVSYLRAKSFFDVALKSSPTERNGGQDNCRAANFREGLNLQEGEFLLQALNGFVLVVTTDALVFYASS
TIQDYLGFQQSDVIHQSVYELIHTEDRAEFQRQLHWALNPSQCTESGQGIEEATGLPQTVVCYNPDQIPP
ENSPLMERCFICRLRCLLDNSSGFLAMNFQGKLKYLHGQKKKGKDGSILPPQLALFAIATPLQPPSILEI
RTKNFIFRTKHKLDFTPIGCDAKGRIVLGYTEAELCTRGSGYQFIHAADMLYCAESHIRMIKTGESGMIV
FRLLTKNNRWTWVQSNARLLYKNGRPDYIIVTQRPLTDEEGTEHLRKRNTKLPFMFTTGEAVLYEATNPF
PAIMDPLPLRTKNGTSGKDSATTSTLSKDSLNPSSLLAAMMQQDESIYLYPASSTSSTAPFENNFFNESM
NECRNWQDNTAPMGNDTILKHEQIDQPQDVNSFAGGHPGLFQDSKNSDLYSIMKNLGIDFEDIRHMQNEK
FFRNDFSGEVDFRDIDLTDEILTYVQDSLSKSPFIPSDYQQQQSLALNSSCMVQEHLHLEQQQQHHQKQV
VVEPQQQLCQKMKHMQVNGMFENWNSNQFVPFNCPQQDPQQYNVFTDLHGISQEFPYKSEMDSMPYTQNF
ISCNQPVLPQHSKCTELDYPMGSFEPSPYPTTSSLEDFVTCLQLPENQKHGLNPQSAIITPQTCYAGAVS
MYQCQPEPQHTHVGQMQYNPVLPGQQAFLNKFQNGVLNETYPAELNNINNTQTTTHLQPLHHPSEARPFP
DLTSSGFL
ONLINE MULTIPLE ALIGNMENT ANALYSIS TOOLS
CINEMA - color interactive editor for multiple alignments
Protein scan against a Profile database at ProfileScan at ISREC
MUSCA
- multiple alignments constrained by sequence motif identification
Tcoffee@igs
- computation, evaluation and combination of multiple sequence alignments.
Examples:
1. For the set of sequences below (first put into
FASTA multiple alignment format):
1. SHKQIYYSDKYDDEEFEYRHVMLPKDIAKLVPKTHLMSESEWRNLGVQQSQGWVHYMIHEPEPHILLFRRPLP
2 MSKDIYYSDKYYDEQFEYRHVVLPKELVKMVPKTHLMTEAEWRSIGVQQSRGWIHYMIHKPEPHILLFRRPKT
3. LQCKILYSDKYYDDMFEYRHVILPKDLARLVPTSRLMSEMEWRQLGVQQSQGWVHYMIHKPEPHVLLFKRPRT
4. PRDTIQYSEKYYDDKFEYRHVILPPDVAKEIPKNRLLSEGEWRGLGVQQSQGWVHYALHRPEPHILLFRRE
5. GQIQYSEKYFDDTFEYRHVVLPPEVAKLLPKNRLLSENEWRAIGVQQSRGWVHYAVHRPEPHIMLFRRPLN
6. GNNDFYYSNKYEDDEFEYRHVHVTKDVSKLIPKNRLMSETEWRSLGIQQSPGWMHYMIHGPERHVLLFRRPLA
7. FIDQIHYSPRYADDEYEYRHVMLPKAMLKAIPTDYFNPETGTLRILQEEEWRGLGITQSLGWEMYEVHVPEPHILLFKREKD
i) Make blocks of the common motif(s) using Block Maker at the BLOCKS server
ii) Find the Block corresponding to yours in the BLOCKS database using LAMA
iii) Generate a phylogenetic tree of the Block sequences from the Block Maker result page
(also at EBI or Pfam)
iv) Search the nr protein database with the Block using MAST
v) Generate a Sequence Logo using the Blocks Multiple Alignment Processor
iv) Identify motifs using MEME and compare
to those obtained with Block Maker
What family of proteins do the sequences above belong to?
2. Identify the conserved domain in sequence 1 above using protein-protein BLAST, followed by a CD Search.
3. Using sequence 1 above as query find other homologs using a HMM-based method at PFAM Protein Search
4. Find information on the domain structure, sequence
and phylogenetic relationships of the Transforming Growth factor beta protein
family
using PFAM and links
to other related resources. Check the results with those found in the previous
section on
5. Identify the motif in sequence 7 above using eMOTIF-SEARCH,
and other sequences that contain it.